- The two chair conformers of substituted cyclohexane have different stabilities.
- Substituents tend to be more stable in the equatorial position than the axial position, since this minimizes steric interactions (1,3-diaxial interactions).
- As substituents become bulkier, they are increasingly unstable in the axial position.
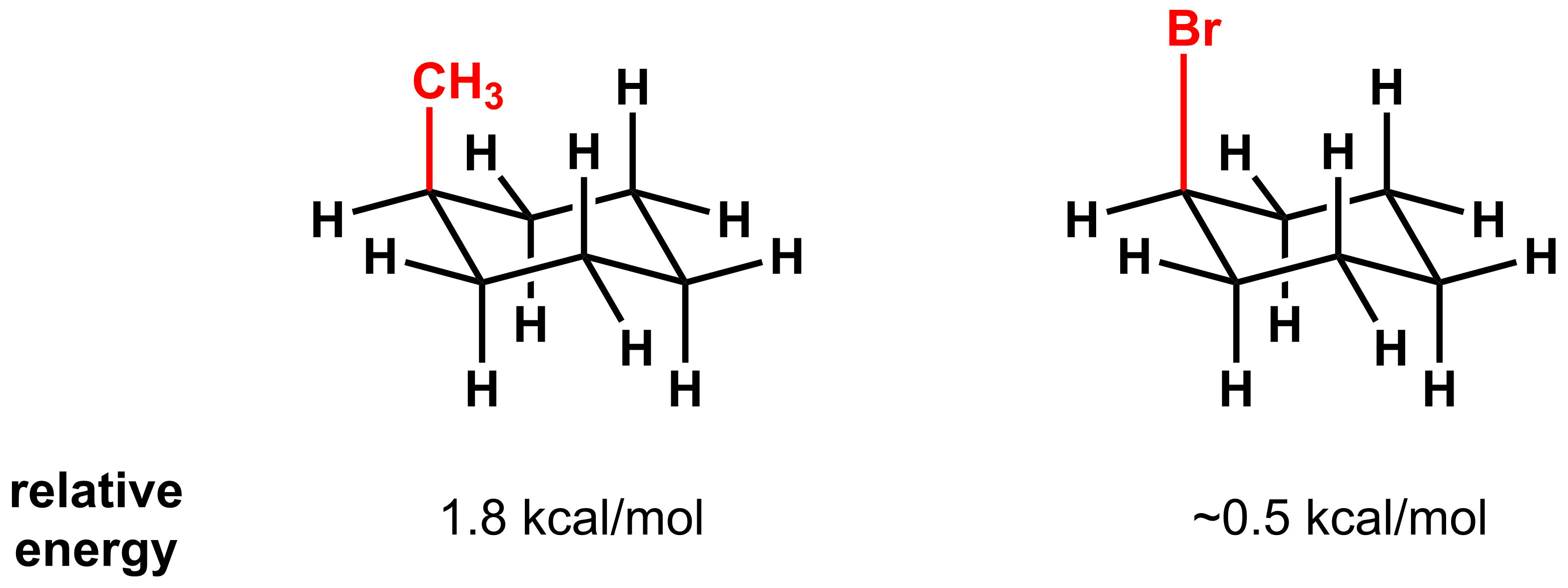
- Substituents with longer bonds experience less 1,3-diaxial interaction.
There is no difference in energy between the two chair conformations of an unsubstituted cyclohexane. However, things become more complicated when we examine the conformations of methyl cyclohexane derivatives. In the example below, the methyl group is on the left-most carbon atom (C1) in an equatorial “up” position. After the chair flip, the methyl group is situated on the left-most carbon in the axial “up” position.
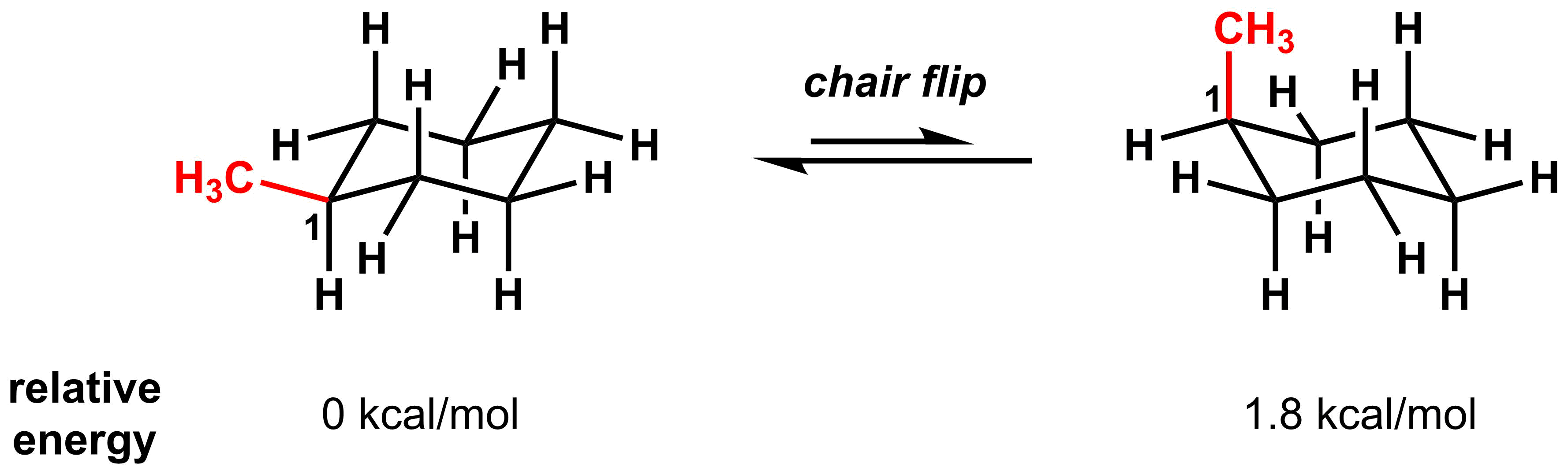
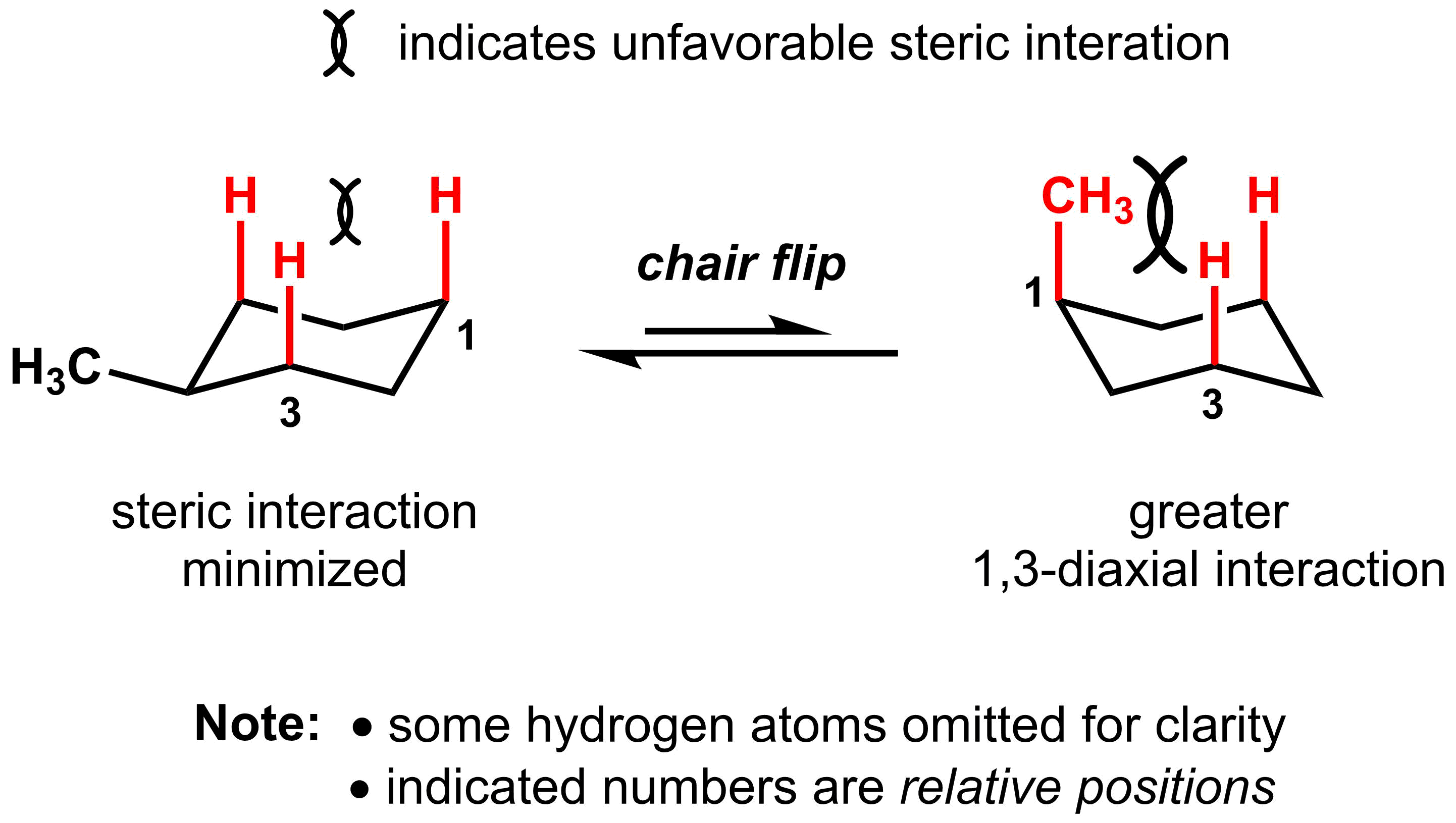
Energetically, these two conformations are no longer equivalent and the conformation with the methyl group in the axial position is 1.8 kcal/mol higher in energy than the conformation with the methyl group in the equatorial position. The reason for the difference is a steric interaction between the hydrogen on the methyl group and the two axial hydrogens on the same face of the cyclohexane. This steric interaction is called a 1,3-diaxial interaction, named for the relative position of the two axial groups. These interactions are not present when the methyl group is in the equatorial position.
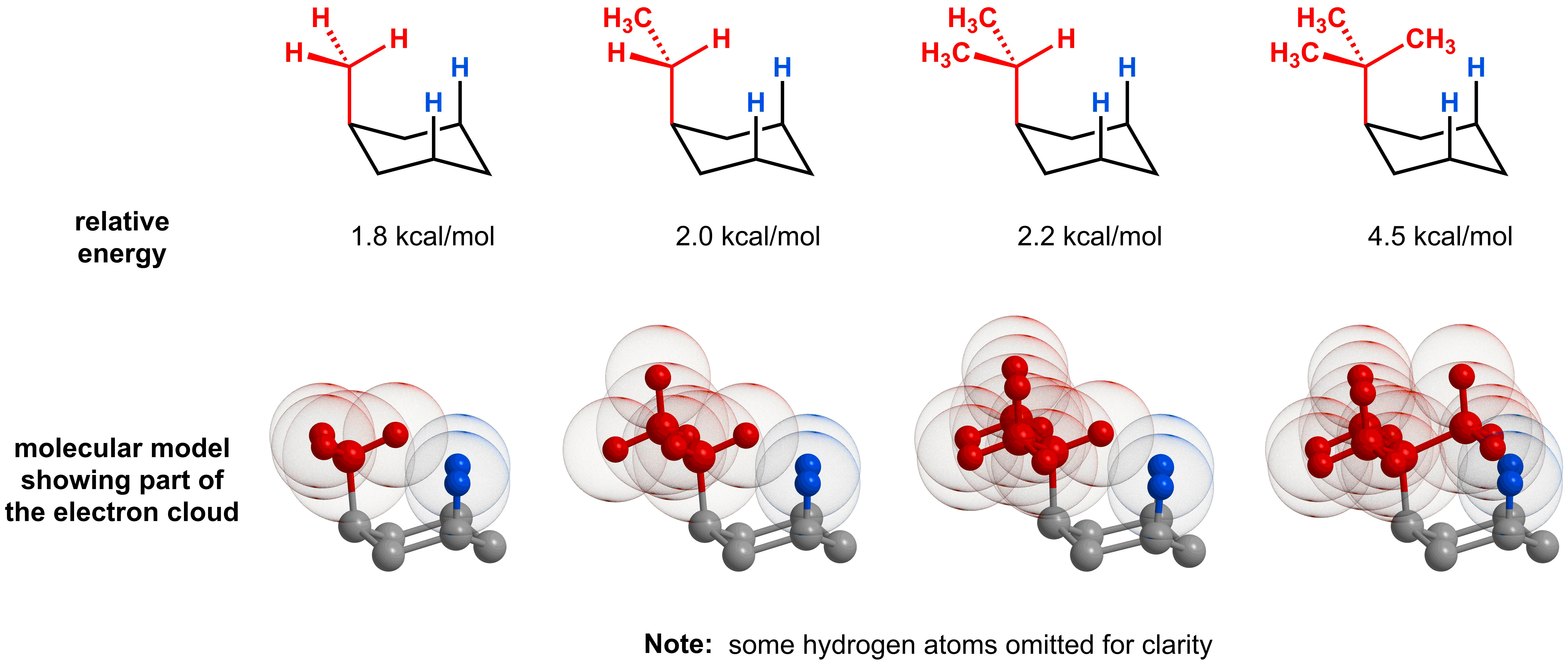
The magnitude of the 1,3-diaxial interaction will be influenced by two key factors. The first factor is the steric size of the subsitutent closest to the ring. As you can imagine, the larger the substituent, the larger the 1,3-diaxial interaction and the higher energy the axial conformer will be. For example, as you increase the substitution close to the ring, from methyl to ethyl, to isopropyl, to tertbutyl, the energy increases. This is because the added branching leads to fewer possibilities to minimize 1,3-diaxial interactions.
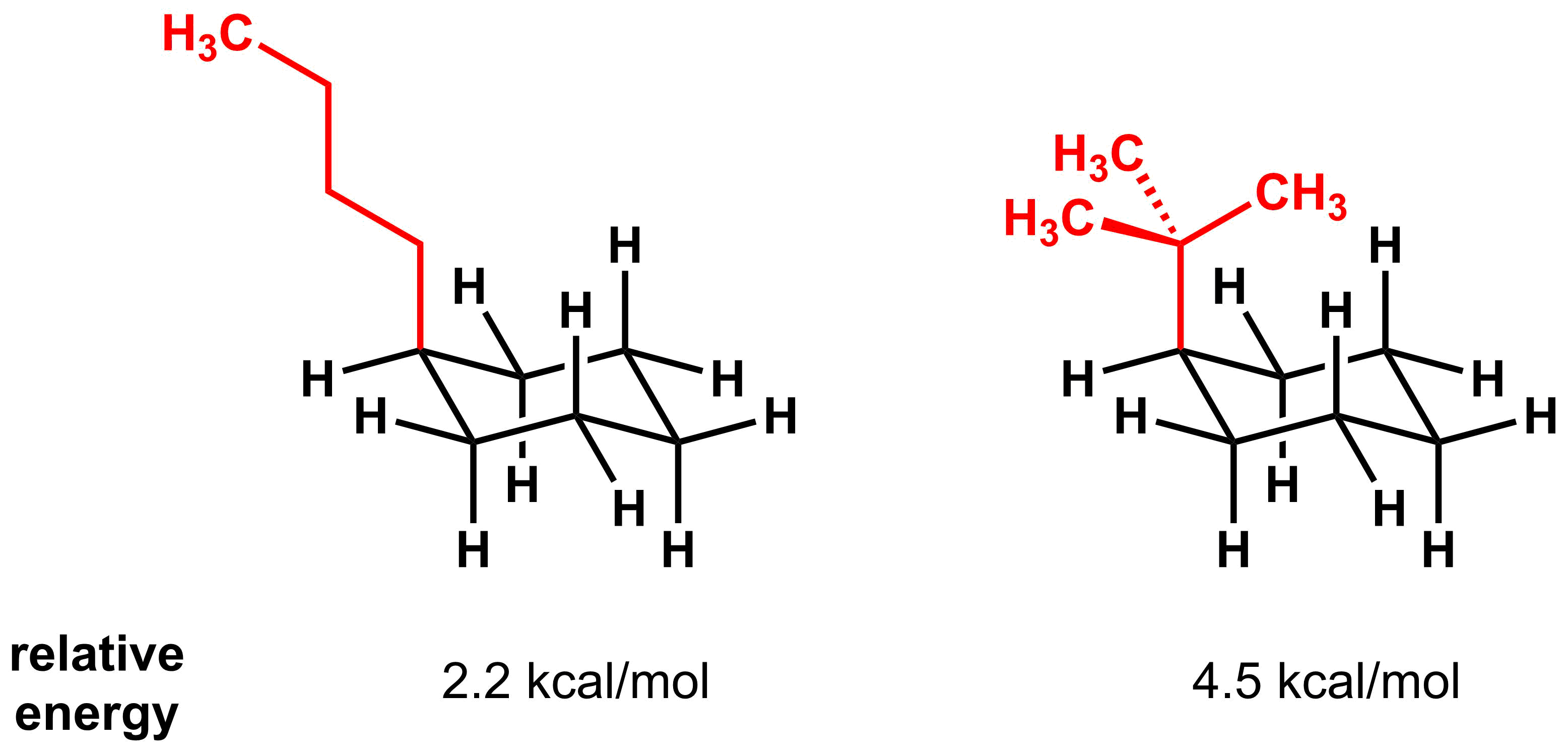
As previously mentioned, one of the key factors is the size of the substituent closest to the ring. For example, if you compare two 4-carbon substituents, the energy of the tert-butyl derivative is significantly higher than the n-butyl derivative because most of the steric bulk of the former is closer to the axial hydrogens, giving few opportunities to minimize the 1,3-diaxial interaction. Even though the n-butyl group has the same molecular weight, most of the steric bulk is far above the axial hydrogens, thus minimizing the steric interaction.
The second important factor in 1,3-diaxial interactions is the bond length of the axial substituent. This can be seen in the 1,3-diaxial interactions in methyl cyclohexane and bromocyclohexane. The bromine substituent is sterically larger than a methyl group, but the 1,3-diaxial interaction is smaller. The reason for this comes down to bond length. Carbon-bromine bonds are longer (194 pm) than carbon-carbon bonds (154 pm), and therefore the steric radii of the bromine is farther from the two axial hydrogens, thus minimizing the steric interaction.
Note: the interactive components of this tutorial require html5 video, which is not supported by some mobile devices (e.g. iPhones). This tutorial is best viewed on a computer.
Interactive: